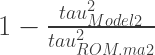I really like meta-analysis, as some of you might already know.
It has it’s critics but used well it can help us achieve one of the major goals of applied ecology: generalisation. This way we can say how we think things generally work and draw conclusions bigger than individual studies.
However, for some reason people have got it into their head that meta-analysis is hard. It’s not. The hardest thing is the same as always, coming up with a good question/problem.
However, for a first timer it can seem daunting. Luckily there are a few good R packages for doing meta-analysis, my favourite of which is metafor.
The aim of these posts is to take some of the mystery out of how to do this and was prompted by a comment from Jarret Byrnes on a previous blog post.
In the first post I will deal with effect sizes, basic meta-analysis, how to draw a few plots and some analyses to explore bias. In the second I will go through meta-regression/sub-group analysis and a few more plots. It shouldn’t be too hard to understand.
For this I will use a dataset based on some work I am doing at the moment looking at the effects of logging on forest species richness. However, I don’t really feel like sharing unpublished data on the web so I’ve made some up based on my hypotheses prior to doing the work.
First of all we need to install the right packages and open them
install.packages("metafor")
install.packages("ggplot2")
library(metafor)
library(ggplot2)
Next we need the data, download it from here and get r to load it up:
logging<-read.csv("C:whereveryouputthefile/logging.csv")
This file contains data on species richness of unlogged (G1) and logged (G2) forests, it’s not perfect as all of the data is made up – it should be useful for an exercise though.
First we need to calculate the effect size. This is essentially a measure of the difference between a treatment and control group, in our case logged (treatment) and unlogged (control) forests. We can do this in different ways, the most commonly used in ecology are the standardised mean difference (SMD) and log mean ratios (lnRR).
The SMD is calculated like this:
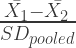
where 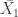 and
and  are the different group means,
are the different group means, 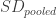 is the pooled standard deviation ( I won’t explain this here but there are good explanations elsewhere). Essentially what this does is to give an effect size based on the difference between groups with more imprecise studies having a smaller effect size.
is the pooled standard deviation ( I won’t explain this here but there are good explanations elsewhere). Essentially what this does is to give an effect size based on the difference between groups with more imprecise studies having a smaller effect size.
The lnRR is calculated as:

which gives the log of the proportional difference between the groups. I quite like this as it is easier to interpret than the SMD as the units can be back-transformed to percentages.
In metafor you calculate SMD by doing this:
SMD<-escalc(data=logging,measure="SMD",m2i=G1,sd2i=G1SD,n2i=G1SS,m1i=G2,sd1i=G2SD,n1i=G2SS,append=T)
and lnRR like this:
ROM<-escalc(data=logging,measure="ROM",m2i=G1,sd2i=G1SD,n2i=G1SS,m1i=G2,sd1i=G2SD,n1i=G2SS,append=T)
From now on in this post I will use the lnRR effect size.
Next we carry out a meta-analysis using the variability and effects sizes of each set of comparisons to calculate an overall mean effect size, using the idea that we want to give most weight to the most precise studies.
In metafor you do that by doing this:
SMD.ma<-rma.uni(yi,vi,method="REML",data=ROM)
summary(SMD.ma)
This is the code for a mixed effects meta-analysis, which basically assumes there are real differences between study effect sizes and then calculates a mean effect size. This seems sensible in ecology where everything is variable, even in field sites which quite close to each other.
The code it spits out can be a bit daunting. it looks like this:
Random-Effects Model (k = 30; tau^2 estimator: REML)
logLik deviance AIC BIC
-27.8498 55.6997 59.6997 62.4343
tau^2 (estimated amount of total heterogeneity): 0.3859 (SE = 0.1044)
tau (square root of estimated tau^2 value): 0.6212
I^2 (total heterogeneity / total variability): 98.79%
H^2 (total variability / sampling variability): 82.36
Test for Heterogeneity:
Q(df = 29) = 2283.8442, p-val < .0001
Model Results:
estimate se zval pval ci.lb ci.ub
-0.4071 0.1151 -3.5363 0.0004 -0.6327 -0.1815 ***
Essentially what it’s saying is that logged forests tend to have lower species richness than unlogged forests (the model results bit), and that there is quite a lot of heterogeneity between studies (the test for heterogeneity bit).
We can calculate the percentage difference between groups by doing taking the estimate part of the model results along with the se and doing this:
(exp(-0.4071)-1)*100
(exp(0.1151*1.96)-1)*100
to get things in percentages. So, now we can say that logged forests have 33% less species than unlogged forests +/- a confidence interval of 25%.
We can summarise this result using a forrest plot in metafor, but this is a bit ugly.
forest.rma(SMD.ma)
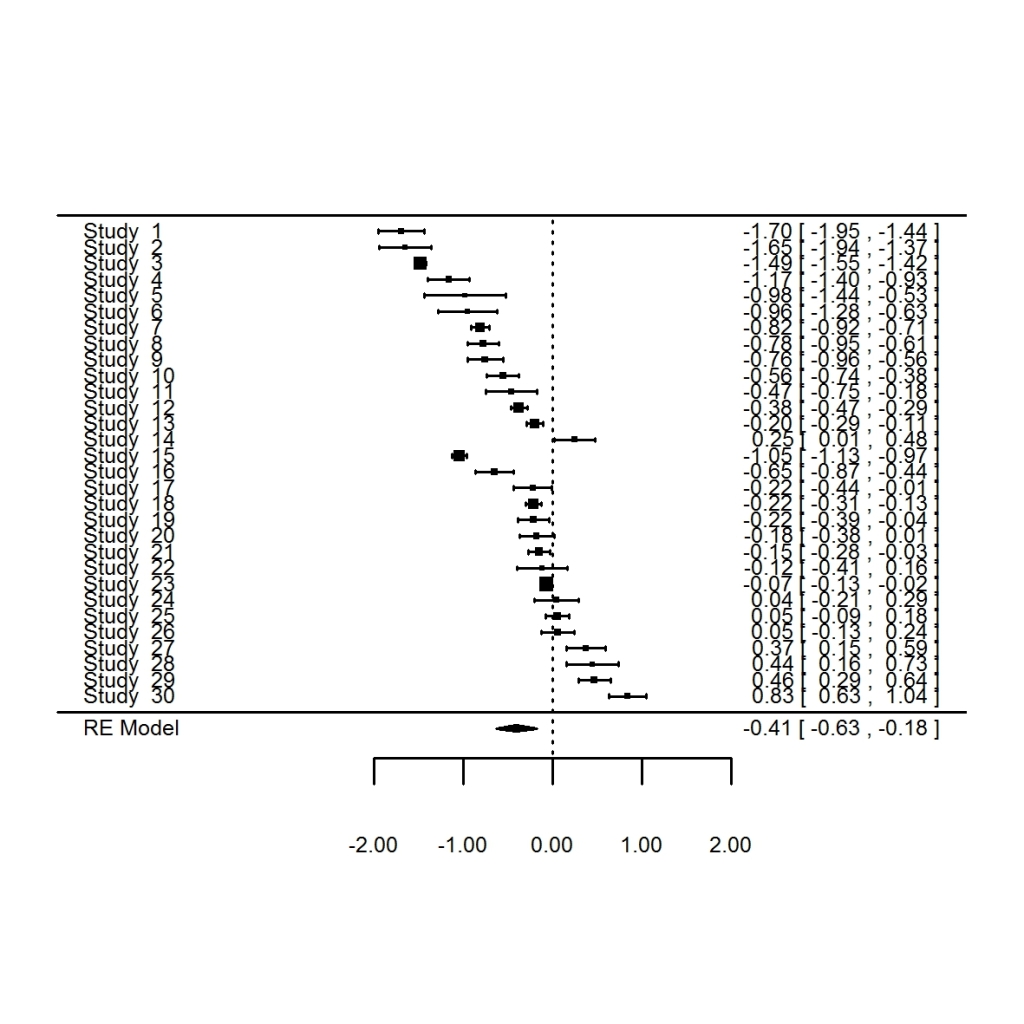
What did I tell you? Pretty ugly right?
Much better to use ggplot2 to do this properly. But this is a bit of a pain in the arse:
theme_set(theme_bw(base_size=10))
forrest_data<-rbind(data.frame(ES=ROM.ma$yi,SE=sqrt(ROM.ma$vi),Type="Study",Study=logging$Study),data.frame(ES=ROM.ma$b,SE=ROM.ma$se,Type="Summary",Study="Summary"))
forrest_data$Study2<-factor(forrest_data$Study, levels=rev(levels(forrest_data$Study)) )
levels(forrest_data$Study2)
plot1<-ggplot(data=forrest_data,aes(x=Study2,y=ES,ymax=ES+(1.96*SE),ymin=ES-(1.96*SE),size=factor(Type),colour=factor(Type)))+geom_pointrange()
plot2<-plot1+coord_flip()+geom_hline(aes(x=0), lty=2,size=1)+scale_size_manual(values=c(0.5,1))
plot3<-plot2+xlab("Study")+ylab("log response ratio")+scale_colour_manual(values=c("grey","black"))
plot3+theme(legend.position="none")

There. Much better.
With a little tweak we can even have it displayed as percentage change, though your confidence intervals will not be symmetrical any more.
theme_set(theme_bw(base_size=10))
plot1<-ggplot(data=forrest_data,aes(x=Study2,y=(exp(ES)-1)*100,ymax=(exp(ES+(1.96*SE))-1)*100,ymin=(exp(ES-(1.96*SE))-1)*100,size=factor(Type),colour=factor(Type)))+geom_pointrange()
plot2<-plot1+coord_flip()+geom_hline(aes(x=0), lty=2,size=1)+scale_size_manual(values=c(0.5,1))
plot3<-plot2+xlab("Study")+ylab("Percentage change in richness\n as a result of logging")+scale_colour_manual(values=c("grey","black"))
plot3+theme(legend.position="none")+ scale_y_continuous(breaks=c(-50,0,50,100,150,200))
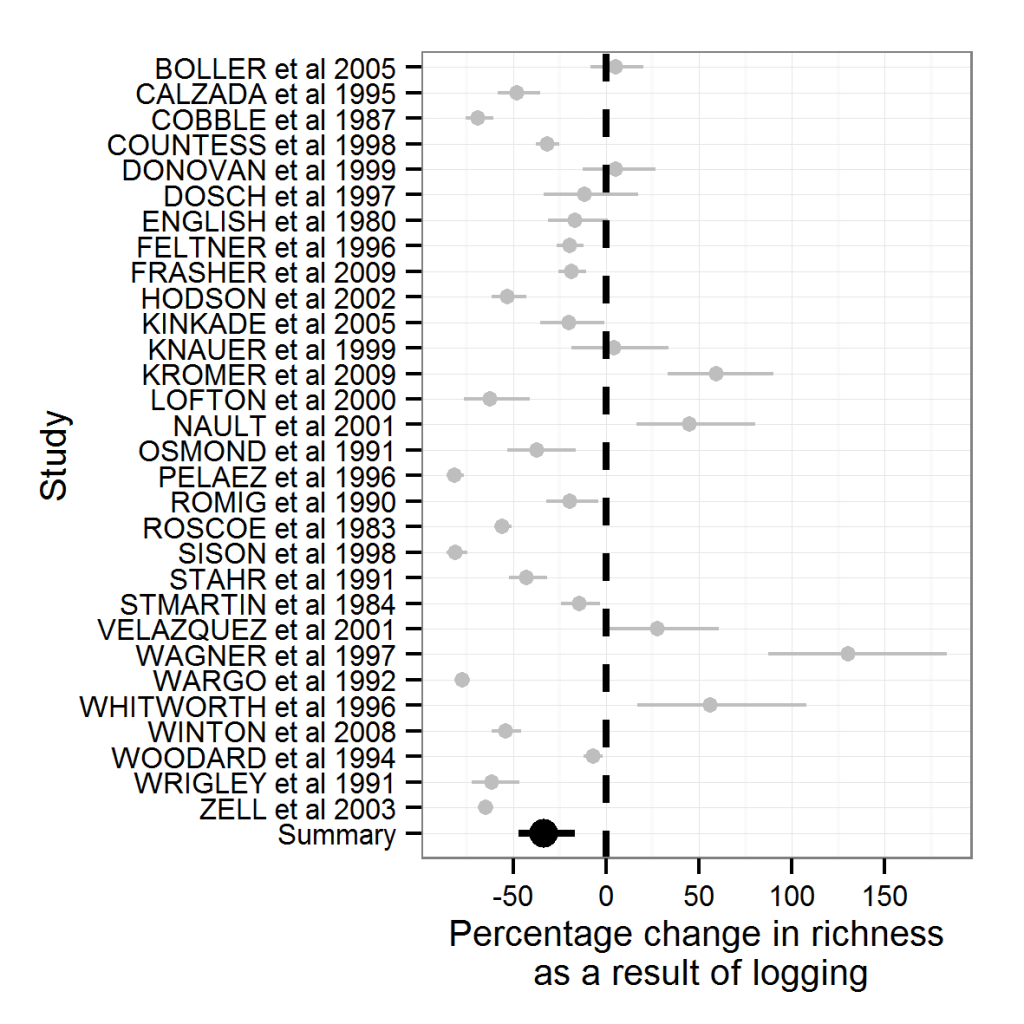
There that’s a bit more intuitively understandable, isn’t it?
Right, I will sort out the next post on meta-regression and sub-group analysis soon.
In the meantime feel free to make any comments you like.